The RTG explores the complex interplay between neurodevelopment/-developmental disorders and adult-onset neuropsychiatric and neurodegenerative diseases. Projects address the following key questions:
 | Research area 1: How do disease genes impact on neurodevelopment? |
| Research area 2: How do neurodevelopmental factors influence vulnerability to disease-precipitating insults? | |
| Research area 3: What are the commonalities in genetics and pathophysiological pathways between neurodevelopmental and neuropsychiatric / -degenerative diseases? |
The RTG investigates these questions from the molecular to the behavioral level in animal models, hiPSC-based cellular model systems and patient-specific biomaterials using state-of-the-art technologies in human genetics, genome-editing, imaging, animal behavior, electrophysiology, pharmacology, developmental and molecular biology, and biochemistry.
In the following three doctoral researchers give you a short insight into their projects.
Marie-Theres Wittmann

Q: Marie-Theres, when did you join the GRK2162?
I studied Molecular Medicine at the University of Erlangen and joined the GRK in 2017 as a PhD researcher in the group of Professor André Reis at the Institute of Human Genetics.
Q: What is the topic of your PhD thesis work?
My project focuses on the transcriptional networks controlling the development of the Corpus Callosum. The Corpus Callosum is the main fibre tract connecting the two hemispheres of the brain. Disruption of its formation is associated with a number of neurodevelopmental and neuropsychiatric disorders. I aim to understand how TCF4, a factor associated to neurodevelopmental disorders, is involved in the formation of the corpus callosum.
Q: How do you approach this question?
I am studying a knockout mouse model of Tcf4 and analyse it using single-cell RNA-Sequencing combined with histological, molecular and biochemical tools.
Q: What is your most exciting finding?
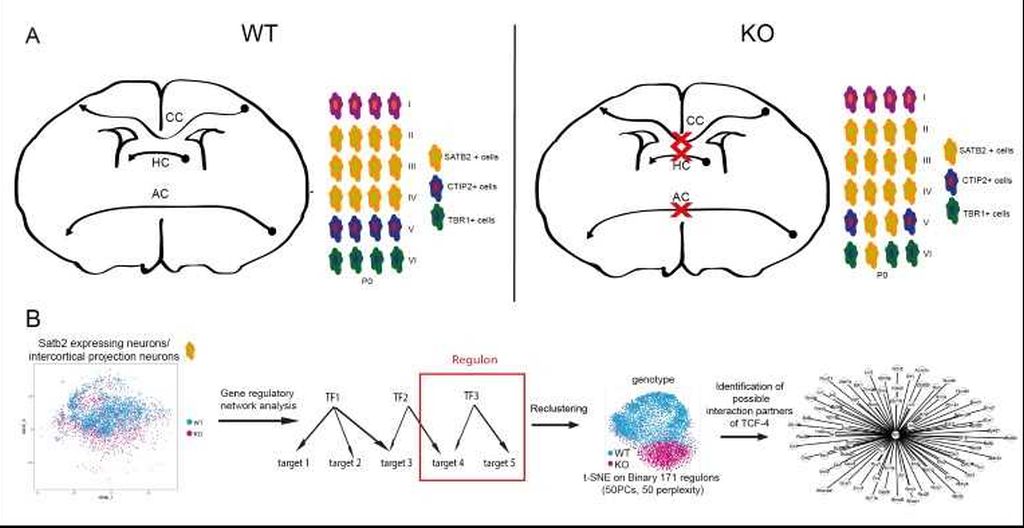
I am very excited about my finding that Tcf4 knockout mice fail to develop all major forebrain commissure. Using single-cell RNA-Sequencing combined with a novel bioinformatical approach I found that TCF-4 orchestrates the activity of a transcription factor network in corpus callosum development. A particularly interesting finding was that this network comprises a number of factors that have been linked to the pathogenesis of neurodevelopmental and neuropsychiatric disorders.
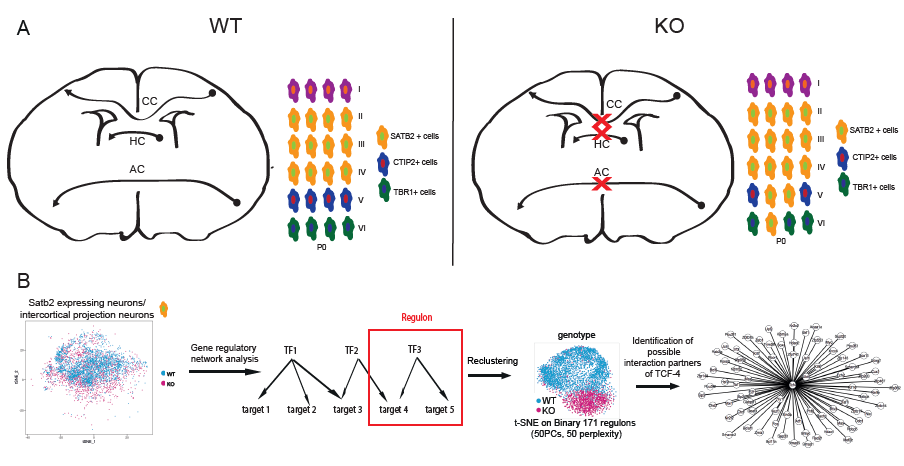
Summary: TCF-4 controls the development of the main fibre tracts of the forebrain. A) Short summary of the results of the histological analysis of the Tcf4 knockout mice: No commissure formation is apparent in the KO mice and the layer formation is dysbalanced in favor of upper layer neuron formation. B) Single cell RNA-Sequencing data of Satb2 expressing cells was used for gene regulatory network analysis using SCENIC. On the basis of co-expression of transcription factors and target genes the activity of the transcription factor regulon was determined. Cells were clustered according to regulon activity and a divergence between WT and KO cells was observable with regulons being less active in the KO. By integrating differential gene expression analysis, potential interaction partners of TCF-4 were identified.
Tatyana Pozner

Q: When did you join the GRK2162?
After finishing my bachelor in neuroscience and then a master thesis in medical sciences in Israel I came to Germany to join the group of Prof. Beate Winner at the Department of Stem Cell Biology. Being always interested in neuroscience I was very enthusiastic to join the GRK2162 when starting my PhD in September 2016.
Q: What is the topic of your PhD thesis work?
I am working on understanding the mechanisms involved in the pathology of a rare motor neuron disorder – Hereditary Spastic Paraplegia Type 11. The patients present with both neurodevelopmental and neurodegenerative deficits, such as thin-corpus callosum and motor neuron degeneration. These deficits lead to learning disabilities and walking difficulties. Currently, no cure exists for the disease.
The mutated gene, SPG11, leads to loss of function of a large protein named Spatacsin. The function of this protein is not well understood. I aim to expand the existing knowledge in a manner that will have clinical significance.
Q: What is the experimental approach?
The approach has several main components. First of all, I look at the phenotype of the disease by using patient iPSC and differentiating them into 2D and 3D neuronal cultures. Then, I try to understand the involved mechanism by using genome editing technology. For instance, because there are no specific antibodies for the protein (spatacsin), its function cannot be reliably studied. We have therefore used CRISPR/Cas9 genome editing technology to tag SPG11, and thus, to overcome this issue. Then we proceed with molecular and biochemical assays. Finally, we treat the cells with a compound that is involved in the modulation of the dysregulated pathways of the disease, to see whether we can rescue the observed phenotypes.
Q: What is your most exciting finding?
I am currently in the process of validating my most exciting finding, i.e., linking the disease to a broader spectrum of neurodegenerative disorders, so I will discuss my second most exciting finding. We were able to identify premature neurogenesis as the main neurodevelopmental deficit at the core of the disorder. This deficit leads to cell-death prone neurons that present with reduced neurite complexity and membranous inclusions. What is most interesting, is that tideglusib, a compound that received FDA fast track status, is capable to rescue all of these defects, thus paving a path for potential clinical application.
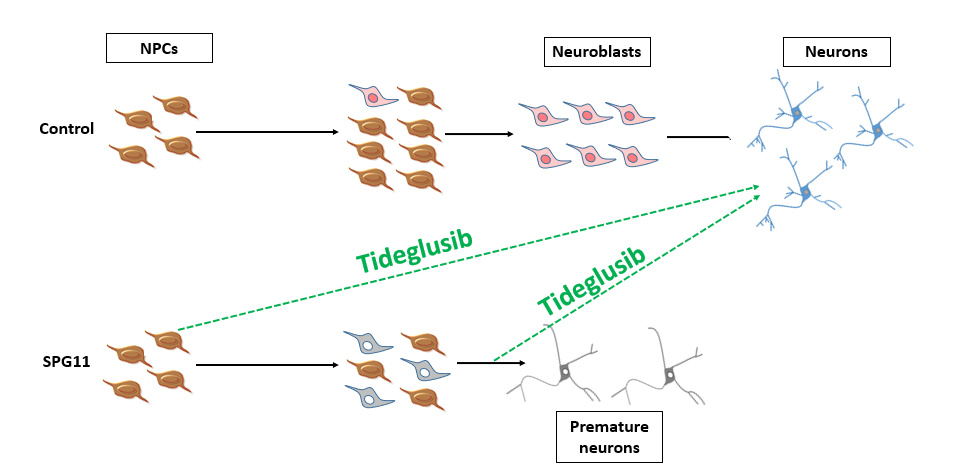
Summary: The control neural progenitor cells (NPCs) give rise to neuroblasts that are eventually differentiated into healthy cortical neurons. In Hereditary Spastic Paraplegia type 11, as a result of an increased rate of asymmetric divisions, neuroblasts are generated at an earlier time point and exhibit less proliferation. Eventually, these neuroblasts differentiate into death-prone neurons with impaired neurite-morphology. The addition of tideglusib at different differentiation stages is capable to rescue most of the observed deficits (adapted from Perez-Branguli, Buchsbaum, Pozner et al. 2018; Pozner, Schray et al. 2018).
Jan Wittstatt

Q: When did you join the GRK2162?
I joined the GRK as a PhD student of Dr. Reiprich at the Institute of Biochemistry in the fall of 2016, just after my master thesis in Cell and Molecular Biology at the University of Erlangen, and finished my PhD in early spring 2020.
Q: What was the topic of your PhD thesis work?
In my project, I investigated aspects of the developing central nervous system with focus on the oligodendroglial lineage. Oligodendrocytes wrap axons of the central nervous system with myelin sheaths and are thereby essential for rapid neuronal signal transduction. A complex network of transcription factors tightly regulates oligodendrocyte maturation. Especially Sox10, a member of the SoxE group, is crucial in oligodendrogenesis. Recent findings also emphasize the importance of microRNAs during oligodendrocyte development. With my thesis, I aimed to analyse the regulatory relation of Sox10 with microRNAs as well as the corresponding impact on oligodendrogenesis.
Q: How did you approach this question?
I identified Sox10-regulated microRNA miR-204 by correlation of microRNA- and Sox10-expression levels as well as transcriptional studies. Effects of miR-204 overexpression and corresponding target genes were analysed in primary cell culture and cell lines by immunocytochemistry and additional biochemical methods.
Q: What is your most exciting finding?
The most exciting finding is the parallel mode of action of miR-204 on oligodendrocyte proliferation as well as differentiation. The expression of miR-204 depends on the master regulator of oligodendrogenesis Sox10. Thus, my results reveal a new regulatory network, in which Sox10 promotes differentiation and initiates cell-cycle exit through the negative regulation of immaturity factors Sox4 and Ccnd2 (CyclinD2) by miR-204. These findings add further detail to our knowledge of the role of Sox10 during oligodendrogenesis and our understanding of oligodendroglial development in general.
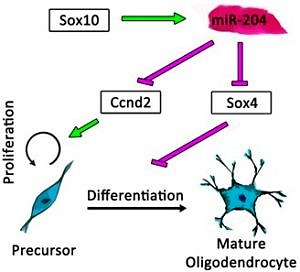
Summary: The function of Sox10-driven miR-204 in oligodendroglial development. This microRNA promotes the differentiation and interferes with proliferation of oligodendroglial cells by negative regulation of Ccnd2 and Sox4 (adapted from Wittstatt et al. 2020).